Plasminogen Deficiency
Disease Overview
Plasminogen deficiency is a disorder that results in unusual and significant clinical sequelae characterized by development of fibrin-rich “pseudomembranes” that impair normal tissue and organ function. The lesions are usually described as woody or ligneous membranes. Ophthalmologic lesions are the hallmark manifestation of plasminogen deficiency and are most frequently reported in the literature; however, other physiologic systems are affected including the gingiva, oropharynx, middle ear, renal collecting system, respiratory tract, central nervous system, skin, and female reproductive tract.1 Poor wound healing has been described.2
Ligneous lesions were first described in 1847 by Bouisson in a young patient who developed pseudomembranes that covered the eye.1 In 1933, the term ligneous conjunctivitis was introduced to describe the wood-like appearance of these pseudomembranes. Thereafter, similar lesions were noted to affect the gingiva, middle ear, respiratory tract, and female reproductive tract.1 In 1958, Verhoeff reviewed the literature after encountering a case of ligneous conjunctivitis and hypothesized that these lesions were due to an infectious agent as they were often reported in association with nasopharyngitis.3 Later, two patients with this condition were thoroughly evaluated for associated infectious and/or autoimmune conditions. When no demonstrable infectious or autoimmune condition could be found, the authors postulated that an inherited etiology was the cause of the ligneous lesions.4
The realization that substances in blood dissolved thrombi occurred in the 19th century; however, isolation of plasminogen/plasmin did not occur until the 1940’s.5 In 1978 a group from Japan were the first to report a case of decreased plasminogen activity with normal antigenic levels (dysplasminogenemia) in a patient with recurrent thrombosis.6
It was not until the 1990s that a clear correlation between decreased plasminogen levels and development of ligneous conjunctivitis was firmly established in a young female with both ligneous conjunctivitis and congenital hydrocephalus.1 Subsequently, two further cases firmly established this relationship.7
While plasmin has a well-established pivotal role in clot dissolution, it also plays other physiologic roles and is critical to normal wound healing. Two mammalian enzymes, tissue-Plasminogen Activator (t-PA) and urokinase-Plasminogen Activator (u-PA), convert the zymogen, plasminogen, to the active enzyme, plasmin. This tightly regulated system is controlled by Plasminogen Activator Inhibitors (PAI’s), which inhibit t-PA and u-PA, and alpha-2-antiplasmin, the main plasmin inhibitor (Figure 1; adapted from Jfdwolff at en.wikipedia).
Figure 1
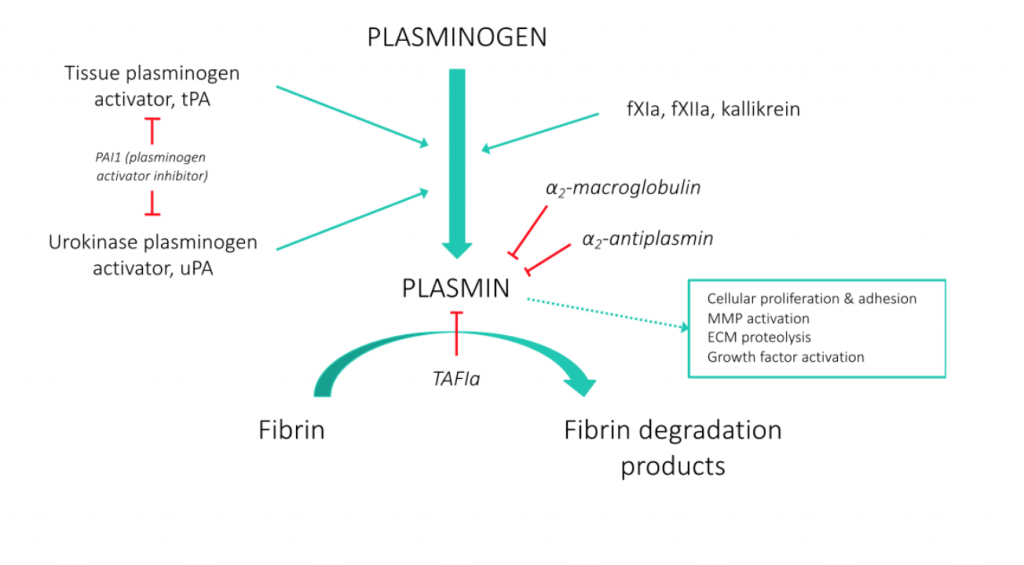
The gene for plasminogen is encoded on the long arm of Chromosome 6, 6q26, and contains 19 exons and 18 introns.8 Plasminogen is predominantly produced in the liver; however, other organs have been documented to produce plasminogen including the brain, kidney, heart, lungs, uterus, spleen, thymus, and intestines.8 The gene encodes for full length plasminogen, referred to as Glu-plasminogen for its N-terminal amino acid. Glu-plasminogen circulates in a closed conformation, restricting access of the plasminogen activators (PAs) to the cleavage site. Upon binding to fibrin or a cell surface, Glu-plasminogen assumes a more open conformation, allowing the PAs access to the cleavage site, in turn producing Glu-plasmin. The initial Glu-plasmin generated cleaves Glu-plasminogen to Lys-plasminogen, which is more stable in the open conformation, facilitating cleavage to plasmin by the PAs.9,10 Total plasma concentration of plasminogen is approximately 200 µg/ml, of which Glu-plasminogen is the dominant form.11 These two forms of plasminogen not only differ in their N-terminal residue and their susceptibility to cleavage by PAs, but also their plasma half-lives, with Glu-Plasminogen having a half-life of 2.2 days versus Lys-plasminogen with half-life of 0.8 days.12
The fibrinolytic system plays an important role in normal wound healing, as it appears to be involved in both initiation of the inflammatory phase, as well as termination of that response.8,9 When a wound is generated, plasminogen levels increase locally, where it stimulates immune cells to release pro-inflammatory cytokines and phagocytose debris.9,13 Furthermore, plasmin aids in the degradation of fibrin within the wound and activates other metalloproteases, which break down the extracellular matrix and aid in tissue remodeling. Finally, it plays a role in slowing inflammation and starting the proliferation phase.9 Therefore, a deficiency of plasminogen results in a diminished capacity to regulate a normal inflammatory response and break down fibrinous material. The inability to progress past this stage in wound healing, particularly in mucous membranes, results in development of the observed ligneous lesions seen in plasminogen deficient patients. Interestingly, plasminogen knockout mice develop similar ligneous conjunctival lesions as those seen in humans.14
Plasminogen deficiency is divided in two main categories, types I and II, both of which are autosomal recessive conditions. Affected patients are either homozygous for a specific mutation or compound heterozygous with each gene having different mutations. Type I deficiency represents a quantitative defect of protein product with decreased plasminogen activity and antigen levels. Several studies have reported numerous mutations within the plasminogen gene that result in a type I deficiency state.15,16 Of these, the K19E mutation appears to be most common; it was found in 17 of 50 subjects in one study, and 6 of 23 subjects in another.15,16 In a Scottish blood donor study, 13 of 15 patients with low plasminogen levels had this K19E mutation.17 This mutation has a variable clinical phenotype with a range of plasminogen levels. Importantly, other mutations do not necessarily confer a predictable clinical expression. One family with the W597C mutation included three siblings, homozygotes for this mutation, where one presented with ligneous gingivitis, one with ligneous conjunctivitis, and one without clinical symptoms.16
Mutations resulting in a dysfunctional protein have been well documented and are associated with type II deficiency.18,19,20,21 The mutations that lead to a dysfunctional plasminogen protein appear more commonly than type I mutations. Heterozygous type I mutations are found in <1% of the populations studied;17,19 however, type II plasminogen gene mutations have been documented with a heterozygous prevalence of 3.8%, 1.5% and 1.6% in separate reports from Japan, China and Korea respectively.20,21 Interestingly, while initial reports suggested that inherited dysplasminogenemia was associated with an increased risk of venous thrombosis, a large population study has not supported this association.20 A recent report on Plasminogen Tochigi knockout mice suggested that there was no increased risk of thrombotic complications, when challenged under experimental thrombotic conditions.22 Plg-Tochigi is the form of dysplasminogenemia that was present in the initial report of this condition in 1978.6 It seems that most people with type II deficiency may be asymptomatic, which might explain the higher prevalence of these mutations.19
A genome wide association study (GWAS) was performed to evaluate genetic variants to determine genetic and environmental contributions in observed variability in plasminogen levels in normal adults.23 This pivotal study revealed four major single nucleotide polymorphisms (SNPs), the forth SNP in linkage disequilibrium with the third, that contribute to plasminogen level variability; in addition, sex (female versus male) and smoking were documented to increase observed plasminogen levels. The SNPs identified were in the PLG and LPA genes located on chromosome 6, and the SIGLEC 14 gene located on chromosome 19. The three most important SNPs identified in this study include:
- rs4252129 in PLG gene resulting in a lowered PLG level
- rs1084651 in an intron of LPA gene resulting in lowered PLG level
- rs10412972 5’ upstream of the SIGLEC 14 gene resulting in increased PLG level
It is not established how these genetic variants impact the expression of plasminogen deficiency in affected individuals.
Plasminogen deficiency is extremely rare; the true prevalence is unknown. A study of blood donors from Scotland revealed 25 of 9,611 tested subjects with a heterozygous plasminogen deficiency.17 Based on these data, a predicted prevalence of homozygous or compound heterozygous plasminogen deficiency would approximate 1.6 per 1,000,000 people.1 Patients from many countries throughout the world have had documented mutations in the plasminogen gene, thereby demonstrating no specific ethnic predilection. In areas where consanguineous or endogamous unions are more common, an increased number of patients have been documented including populations from Turkey and the Middle East. Of 50 patients reported from 11 countries, 21 were of Turkish descent.15 In a subsequent study from this same group, 10 of 23 subjects were of Turkish descent.16
A review of documented cases found an increased number of affected females as compared to males with a ratio of approximately 1.5:1.16 An autosomal recessive inheritance would predict equal distribution among cases; the increased number of identified females may be due to an increased risk of disease expression in females. Clinical age of symptom expression is quite variable, with a median age of 1 year, and a mean of ~5 years.16 However, some patients do not present until much later in life. A recent report of a novel mutation recognized in Japan identified a male who did not present with ligneous conjunctivitis until the age of 58 years. In this case, it appeared that a pulmonary resection was the trigger for him to develop the lesions.24
The lack of longitudinal follow-up and outcome data in plasminogen deficient patients prevents accurate conclusions about the true extent of morbidity and mortality associated with this disorder and the lifelong burden of disease.
