Factor XIII Deficiency
Disease Overview
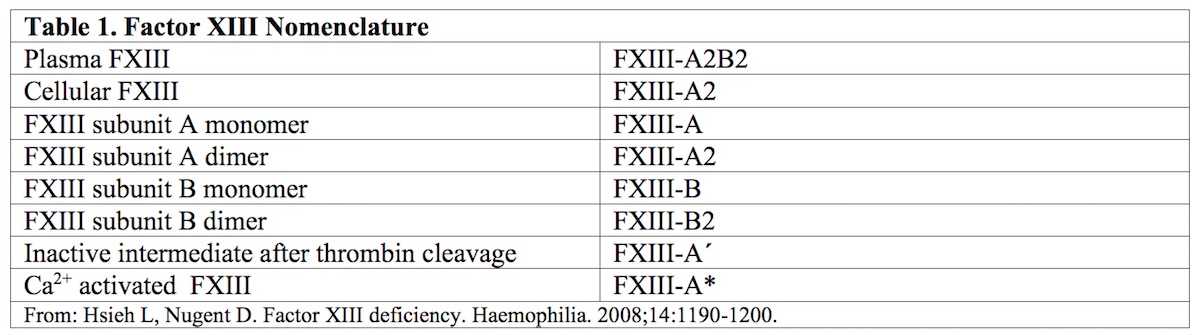
Factor XIII (FXIII) was first identified by Laki and Lorand in 1948, when they surmised and then confirmed, the presence of a clotting factor whose major role was to stabilize and strengthen thrombus formation and to prevent excessive clot breakdown resulting in re-bleeds.1,2 Plasma FXIII circulates as a pro-transglutaminase (FXIII-A2B2) composed of two catalytic A subunits (FXIII-A2) and two non-catalytic B subunits (FXIII-B2) held together by non-covalent bonds. The B subunits are found primarily in plasma, either free or in association with A subunits as part of the heterotetrameric form of FXIII.
There is, in addition, an intracellular form of FXIIIA, which exists only as a homodimer of FXIII-A subunits (FXIII-A2) and primarily plays a role in crosslinking cytoskeletal protein, stabilizing cell-cell interactions, and cellular adhesion to the subendothelial matrix.3
A quick reference for the various forms of FXIII discussed in this article is listed below in Table 1.
Multiple Functions of Factor XIII
Before reviewing the clinical and biochemical details that characterize factor XIII (FXIII) deficiency, it is worth noting that this multifunctional transglutaminase not only crosslinks fibrinogen to stabilize and strengthen clot formation, it also facilitates wound healing, angiogenesis and response to bacterial pathogens. Like so many other discoveries, FXIII is defined by the function through which we first became aware of its existence, namely, as a ‘fibrin stabilizing factor’ (FSF) in the earliest papers.1,2 However, it is now well documented that plasma or circulating FXIII is capable of cross-linking an ever-growing list of proteins critical to the vascular matrix, platelets, endothelial cells, granulocytes and monocytes. Indeed, using a proteomic approach in combination with transglutaminase specific labeling, at least 147 FXIIIa plasma protein substrates were identified, 48 of which were confirmed to be present in clots analyzed in vitro.4
Intracellular FXIII plays an equally important role in the platelet and vascular bed in achieving hemostasis, thrombosis and healing. Although we focus this review article on the interaction of plasma proteins with FXIII, it is clear that intracellular FXIII, especially in the platelet and vascular bed may play an equally important role in hemostasis.
In plasma, FXIII circulates as a pro-transglutaminase (FXIII-A2B2) composed of two catalytic A subunits (FXIII-A2) and two non-catalytic B subunits (FXIII-B2) held together by non-covalent bonds. The B subunits are found primarily in plasma, either free or in association with A subunits as part of the heterotetrameric form of FXIII. However, intracellular FXIII exists only as a homodimer of FXIII-A subunits (FXIII-A2). Please refer to Table 1 for the various forms of FXIII discussed in this article.
Substrates for intracellular FXIII-A2 include myosin, actin, vinculin and filamin, suggesting a major role for FXIII in cytoskeletal remodeling in platelet adhesion, aggregation and contraction. In the monocyte/macrophage cytosol, FXIII-A2 facilitates Fc and complement receptor mediated phagocytosis. Importantly, FXIII deficient patients have been shown to have impaired function in these activities as well.3 Murine models of wound healing also support the important role of intracellular FXIII-A2 in leucocyte and tissue remodeling and repair. Recent research on myocardial repair following cardiac infarction induced in FXIII knockout mice, demonstrates significant reduction in leucocyte recruitment, phagocytosis and protease activity in the area of injury.5 Finally, the production of FXIII-A2 in the placenta, particularly the labyrinthine layer, confirms its important role in maintaining the integrity of placental attachment in the uterus.6-8 This, along with prevention of massive bleeding associated with microvascular bleeds, is no doubt one of the major causes of miscarriage and fetal loss in FXIII deficient women.
Structure and Function
Plasma FXIII, the heterotetramer (FXIII-A2B2), plays an integral role in hemostasis by catalyzing the cross-linking of fibrin, a variety of integrins within the platelet membrane and matrix proteins throughout thrombus formation, thus strengthening and stabilizing the blood clot.
Plasma FXIII is converted from a pro-transglutaminase to activated FXIII (FXIII-A*) by thrombin in the presence of calcium ions and fibrin. FXIII-A contains both the thrombin cleavage site and the calcium-binding site required for catalytic activation.9
The gene coding for the FXIII-A subunit (F13A) is located on chromosome 6p24–25, spanning 160 kb and consists of 15 exons interrupted by 14 introns encoding a mature protein of 731 amino acids.10
FXIII-A is divided into the activation peptide, b-sandwich, catalytic core, b-barrel 1 and b-barrel 2.11 Crystal structure of the A subunit revealed a catalytic triad in the central core domain formed through hydrogen bond interactions between Cys314, His373 and Asp396.12
In the first step of FXIII activation, thrombin cleaves off the FXIII N-terminal activation peptide (AP-FXIII) of 37 amino acid residues at position Arg37, forming the plasma FXIII-A. In the presence of Ca2+ and fibrin, the B subunits then dissociate from the A subunits resulting in a conformational change thus leaving the active site cysteine accessible for the substrate.9
AP-FXIII appears to be responsible for the stable protein expression of the Factor XIII subunit as deletions in the first 11 amino acids result in severe FXIII deficiency.13 Of note, FXIIIA residue at Arg11 appears to be critical in forming the salt bridge to the opposite FXIIIA subunit catalytic core domain residue Arp343 in the stable homodimer.14 This mirror image cross reaction between the activation peptides on one FXIII A subunit and the catalytic domain of the other in the FXIII dimer, promotes stability between the two subunits in keeps the catalytic site concealed while in circulation, until thrombin cleaves the activation peptides away. Recently, Schroeder et al., suggest that once cleaved from FXIIIA subunit, the activation peptide itself (AP-FXIII) also has a down-regulatory effect, reducing FXIII activation and fibrin cross-linking locally to limit thrombus growth.15
Activated FXIII-A2 (FXIII-A2*) then catalyzes the cross-linking of fibrin or other target proteins that contain appropriate glutamine and lysine residues. Activated FXIII covalently cross-links fibrin through a c (c-glutamyl) lysine link. FXIII can cross-link fibrin polymers by c-dimerization, Gln** 398 in one fibrin molecule and Lys406 of the next fibrin molecule.
Polymerization can also occur through the alpha chains of Gln328, Gln366 and Lys508 in multiple fibrin molecules.16 The active site contains cysteine residue (Cys311), which is found within the sequence Tyr-Gly-Gln-Cys-Trp. FXIII also forms a covalent bond with a2-antiplasmin17,18 and thrombin activatable fibrinolysis inhibitor (TAFI), further stabilizing the thrombus, making it more resistant to proteolytic degradation by plasmin.17
Additionally, FXIII also cross-links several other protein substrates in the plasma and subendothelium, including fibronectin, von Willebrand factor, vitronectin, collagen, coagulation factor V, thrombospondin and plasminogen activator inhibitor type 1.9 Thrombin does not play a role in activation of intracellular FXIII, as it does with plasma FXIII. When the intracellular calcium level rises in the presence of target substrate, platelet FXIII undergoes a non-proteolytic conformation change resulting in its active form.19
Role of FXIII-A Dependent Cross-linking in Clot Consolidation
FXIII-A plays a major role in clot formation and consolidation by cross-linking fibrin to produce the structural framework of a thrombus that is able to withstand mechanical breakdown.20
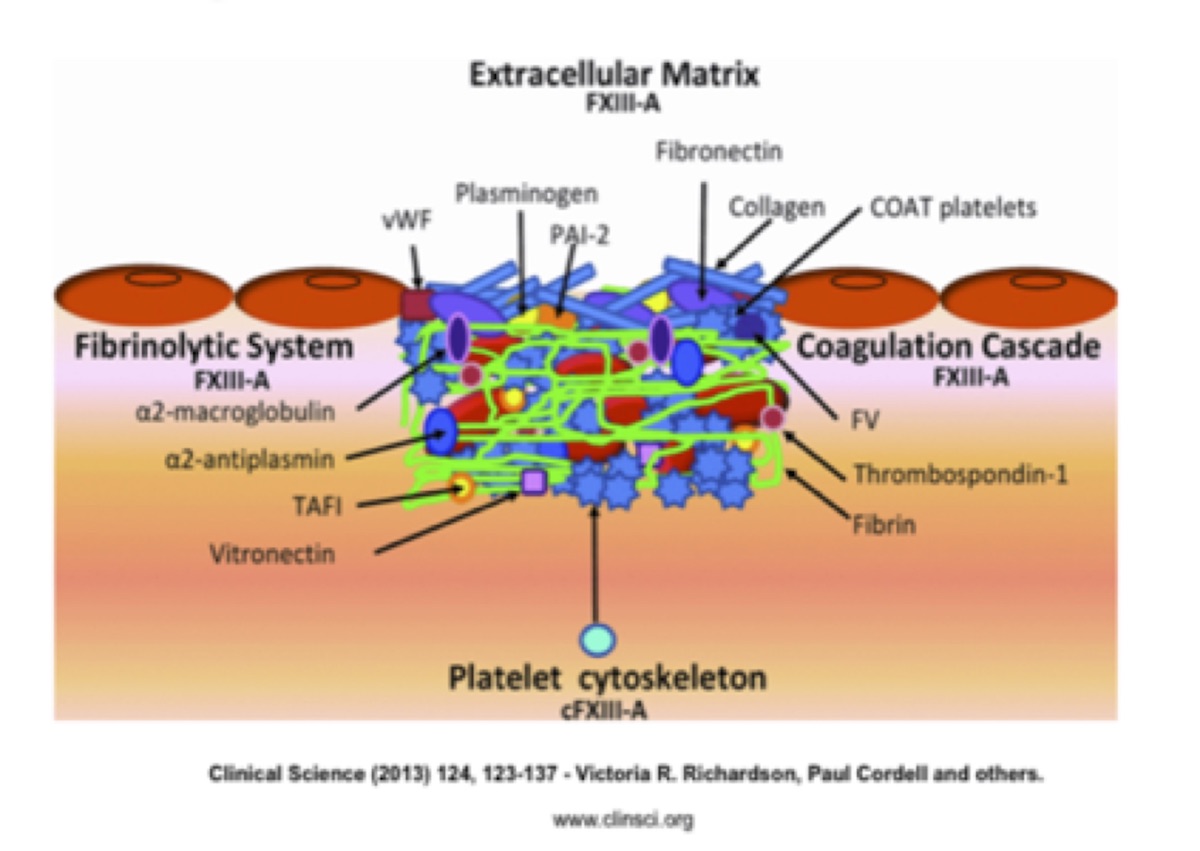
Tissue damage activates the coagulation cascade to generate thrombin. Thrombin cleaves fibrinogen to form fibrin and activates platelets and plasma FXIII-A (Figure 1). The cross-linking of cytoskeleton proteins by intracellular FXIII-A activates platelets to undergo conformational changes. Some form COAT platelets, which are a subset of thrombin and type I and V collagen-activated platelets that have high surface concentrations of α-granule proteins, including FV, fibrinogen, vWF (von Willebrand factor), fibronectin, α2-antiplasmin, and thrombospondin. These platelets aid prothrombinase activity. Plasma FXIII-A cross-links fibrin to produce the structural framework of a thrombus. Cross-linking of the fibrinolysis inhibitors during fibrin formation protects the forming clot from plasmin-mediated lysis.
Cross-linking of matrix proteins fibronectin, collagen and vWF may be important in cell–matrix interactions and cross-linking of PAI-2 and plasminogen to matrix components suggests a role in localized regulation of plasmin generation and matrix metalloproteinase activation.20
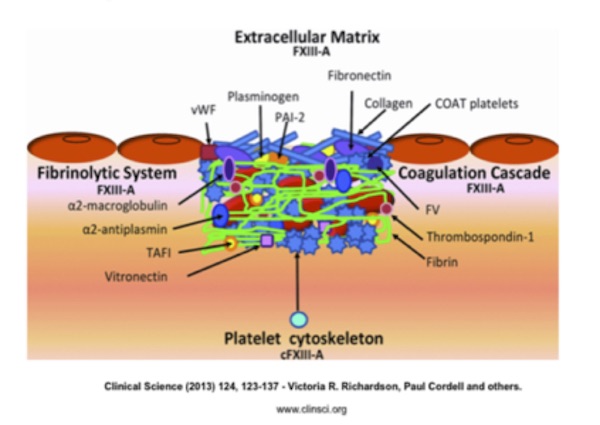
Figure 1. Role of FXIII-A dependent cross-linking in thrombus consolidation and extracellular matrix attachment.
From: Richardson V, Cordell P, Standeven K, Cater A. Substrates of Factor XIII-A: roles in thrombosis and wound healing. Clin Sci. 2013;124:123–137. Used with permission.Synthesis of Factor XIII
The liver is the major site of synthesis for FXIII-B, whereas it appears that the hematopoietic cells are responsible for the production of FXIII-A. Early on, the strongest evidence to support the hepatic origins of the B subunit came from organ and stem cell transplantation. Wall et al. showed that after liver transplantation, the recipient’s FXIII-B phenotype changed to the donor’s phenotype, whereas the FXIII-A remained unchanged. In addition, the FXIII-A phenotype was converted to the donor’s phenotype only after bone marrow transplantation.21 The FXIII A2 is produced in megakaryocytes and monocytes from the earliest stages of myeloid development with circulating platelets containing around 50% of the total FXIII found in whole blood.
Intracellular FXIII-A2 (not FXIII-B) can be found in platelets, megakaryocytes, monocytes, tissue macrophages and placenta. Cellular FXIII-A2 is immunochemically identical to plasma FXIII-A2 and can combine with FXIII-B2 to form the tetramer complex when released into the plasma.
The FXIII-B subunit gene (F13B) is located on chromosome 1q31–32.1 and spans approximately 28 kb in length and is composed of 12 exons interrupted by 11 introns encoding the mature protein of 641 amino acids.22,23 The B subunit of FXIII is composed of 10 tandem repeats called glycoprotein-1 structures or Sushi domains because of their shape. Based on studies by Souri et al., B subunits interact with each other to form dimers through Sushi domains 4 and 9.24
One important function of the B subunit is the stabilization and transport of the hydrophobic A2 subunit in the aqueous environment of human plasma, thereby prolonging FXIII-A2 in circulation. Although there is a growing body of literature describing the role of B subunit in decreasing the degradation and inactivation of the A2 subunit by proteases, its most critical role is the localization of FXIII to the growing fibrin polymer, while the thrombin is still active, to initiate and accelerate the cross-linking process. Localization is facilitated when the FXIII-B portion of the Factor XIII A2B2 molecule binds specifically to the gamma chains of fibrinogen, leading to polymerization, cross-linking and regulation of FXIII activity.25-29
Congenital FXIII deficiency is caused by defects in either FXIII-A subunit genes or FXIII-B subunit genes. The bleeding disorder resulting from mutations in the FXIII B subunit gene is associated with very low levels of circulating B subunit protein, occurs infrequently (< 5% of reported factor XIII deficiency cases) and is less severe in general, as the FXIII A subunit is still produced and remains active in its intracellular form. However, in severe FXIII-A deficiency, the A subunit is absent from all compartments, including plasma, platelets, monocytes and placenta, resulting in a more significant bleeding diathesis and poor wound healing.
The first published genetic mutation leading to FXIII deficiency was reported by Webb et al. in 1992.23 With the advance of molecular and genomic technologies, more than 70 FXIII-A or B subunit gene mutations have now been identified. At the time of this review, 67 mutations were reported for FXIII-A with the vast majority being missense or nonsense mutations. Mutations can occur throughout the gene, but concentrate between exons 3 and 14. A complete listing of all subunit A or B mutations (missense, nonsense, splice, insertions and deletions) can be found at the Human Gene Mutation Database at the website of the Institute of Medical Genetics in Cardiff and at the Factor XIII Registry Database website.
For more details on the mutations for FXIII-B and studies of polymorphism, see Pattern of Inheritance.
The incidence of severe FXIII deficiency is one in 3-5 million people and is inherited in an autosomal recessive pattern.17 This rare bleeding disorder affects people of all races and there is often a history of consanguinity within certain families of FXIII-deficient patients. Among non-consanguineous families, a higher incidence of compound heterozygosity is observed.8,30-32
Prognosis
Although there is a lifelong risk of bleeding with FXIII deficiency, the prognosis is excellent because of the good response to treatment with plasma derived or recombinant Factor XIII concentrates, or blood products such as FFP, cryoprecipitate. Because the half-life of FXIII is long, ranging from 6-10 days, prophylaxis is easily accomplished for those patients even with the worst bleeding history. Unlike with other forms of hemophilia, the incidence of inhibitor development is extremely low.33
