Factor X Deficiency
Disease Overview
Factor X (FX) is a vitamin K-dependent serine protease synthesized in the liver. It plays a key role in coagulation and is the first enzyme in the common pathway that leads to the formation of a stable fibrin clot. Inherited FX deficiency, a rare autosomal recessive bleeding disorder, is estimated to occur in 1:1 000 000 individuals with up to 1:500 being carriers.1
In 1905 Morawitz identified a factor named “thromboplastin” that interacted with “thrombogen” to form thrombin,2 and in 1955 Duckert reported a factor deficiency that was distinct from FVII and FIX deficiencies in patients receiving coumarins.3 Inherited FX deficiency was later identified by 2 independent groups, each of which described a patient with a bleeding diathesis that could not be attributed to deficiencies in other known coagulation factors. The factor in both patients was subsequently named FX.3-5 As with other vitamin K-dependent proteins, FX requires posttranslational carboxylation of 11 glutamic acid (Gla) residues for functional activity. This carboxylation allows for the Ca++-dependent binding of FX to negatively charged phospholipid membranes.
The mature 2-chain form of the FX molecule consists of a light chain of 139 amino acids and a heavy chain of 306 amino acids, linked by a disulfide bond. The light chain contains the Gla domain and 2 epidermal growth factor domains; the heavy chain contains the catalytic serine protease domain. The 59-kDa two-chain protein circulates in the plasma at a concentration of 10 µg/mL.
Activated FX (FXa) is a catalytic serine protease that is generated following cleavage of the heavy chain of FX, releasing the 52-residue activation peptide that contains the His236, Asp228, and Ser379 catalytic site. Both the extrinsic and intrinsic pathways can lead to FX activation: in the extrinsic pathway, this activation occurs via formation of the tissue factor:factor VIIa (TF:FVIIa) complex with calcium ions on a phospholipid surface; in the intrinsic pathway, FX activation occurs when the serine protease factor IXa (FIXa) and its cofactor factor VIIIa (FVIIIa) form the “tenase” complex in the presence of calcium ions on a phospholipid surface.
FXa is the most important activator of prothrombin, cleaving prothrombin to generate thrombin in a complex with factor Va (FVa), Ca ions, and phospholipids. FXa also activates FV and FVIII and hydrolyzes FVII to FVIIa, completing a FVII-FX feedback loop. FXa also forms a quaternary complex with TF:FVIIa by binding to tissue factor pathway inhibitor (TFPI). This process down regulates the extrinsic pathway, reducing thrombin generation.
Antithrombin inactivates FXa by forming a complex that is rapidly cleared from circulation. The protein Z–dependent protease inhibitor, a serine protease inhibitor (serpin) also inactivates FXa. The affinity of this protease inhibitor for FXa is increased 1000-fold by the presence of protein Z.6 Defects in protein Z lead to increased FXa activity and may increase the risk of thrombosis.7
Inherited FX deficiency is a rare autosomal recessive disorder caused by mutations of the FX (F10) gene that results in reduced or aberrant function of the FX protein. More than 130 mutations of the F10 gene have been identified, most of which are missense mutations (Figure 1).
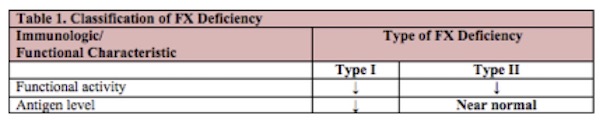
The classification of FX deficiency is based on the results of both immunologic and functional laboratory assays. In patients with type I deficiency, both functional activity and antigen level are proportionally decreased. In type II deficiency, the FX protein is dysfunctional, resulting in reduced FX activity with a near-normal antigenic level (see Table 1).
See the section on Laboratory Evaluation for a proposed classification schema for clinical purposes that is based on clotting, chromogenic, and immunologic characteristics.
FX deficiency is estimated to occur in 1 in 1,000,000 individuals worldwide.1 In populations where consanguinity is more common, the frequency is reported to be 1 in 200,000 individuals. The prevalence of the carrier state is estimated to be 1 in 500 individuals.1 According to the Rare Bleeding Disorders Database survey, patients with FX deficiency represent 10% of the total number of patients affected worldwide by rare bleeding disorders.8
