Congenital Platelet Function Disorders
Platelet Defects
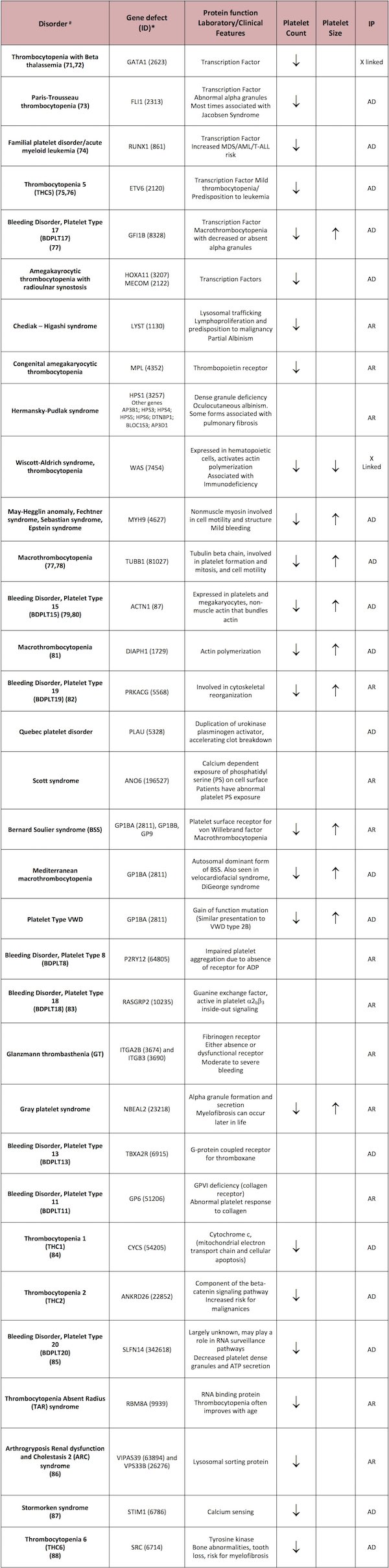
Some disorders will be described in this section. For a complete list of disorders please refer to Table 1.
Microthrombocytopenia (small platelets with thrombocytopenia)
Two forms of thrombocytopenia are characterized by small platelet size: Wiskott-Aldrich syndrome (WAS) and X-linked thrombocytopenia (XLT). Both of these syndromes result from mutations in the WAS gene, but boys with isolated thrombocytopenia who do not exhibit the associated immune disorders seen in WAS have been given the diagnosis of X-linked thrombocytopenia to distinguish them from the more severe disorder historically.
In males, WAS is the most likely diagnosis, which can be confirmed by the presence of WAS protein (WASP) gene variants, cellular and humoral immunodeficiency, eczema, and recurrent infections. Megakaryocytes on bone marrow (BM) studies are typically normal. WAS results from mutations of the WAS gene, encoding WASP, which plays a critical role in actin cytoskeleton organization and signaling. In 77 families with WAS gene variants, gene sequencing identified 62 mutations, including 17 novel sequence variants.20
Initially thought to be isolated to only exon 2 of the WAS gene, it is clear that XLT mutations can occur in other exons, and the main factor that distinguishes XLT from full blown WAS is varying amounts of residual expression of the WAS protein in lymphocytes and platelets.
In XLT, boys are often misdiagnosed with chronic immune thrombocytopenia (ITP) that has been refractory to withdrawal of therapy. Although steroids may be effective in increasing the platelet count, this is not maintained when the medication is discontinued. The platelet size may or may not be informative in this setting, but certainly a decrease in platelet size, mild or intermittent eczema, and a family history of thrombocytopenia on the maternal side of the family should prompt a work-up for possible mutations in the WAS gene.
As the platelets are small, and not the young and more thrombotic form seen with high platelet turnover, these patients often exhibit more bleeding than that seen in a patient with ITP at any given platelet count. In addition, depending on the degree of lyonization, the mother of an XLT patient may also have a history of occasional thrombocytopenia (counts <100,000). Proper diagnosis will often save patients from splenectomy and a complete immunologic evaluation should always be performed, because many of the patients diagnosed with XLT early in life do eventually manifest with immune dysfunction, poor response to immunizations, recurrent infections, or inflammatory autoimmune disorders over time. All XLT patients should be followed-up by specialists who can monitor their immune status closely.
Normothrombocytopenia (normal-sized platelets with thrombocytopenia)
Once it is confirmed that there is no evidence of an immune basis for the thrombocytopenia, normal platelet size may indicate a defect of megakaryopoiesis. Transient thrombocytopenia in neonates is common, but severe, persistent thrombocytopenia suggests a rare inherited condition, such as congenital amegakaryocytic thrombocytopenia (CAMT), amegakaryocytic thrombocytopenia with radio/ulnar synostosis (ATRUS), thrombocytopenia absent radius (TAR), and Paris Trousseau/Jacobsen syndrome (PT/J).
Elevated plasma thrombopoietin (TPO) levels differentiate these disorders from acquired thrombocytopenia due to perinatal or prenatal factors, particularly in CAMT, where plasma TPO levels can be 10-fold higher than normal. ATRUS and TAR both present with characteristic bony abnormalities, including radioulnar synostosis and/or absent radii, based on upper extremity X-ray examination.
Exome sequencing of pedigrees of 53 cases determined that the compound inheritance of a low-frequency regulatory single-nucleotide polymorphism (SNP) and a rare null mutation in the RBM8A gene causes TAR syndrome.21 CAMT is characterized by genetic defects in the TPO receptor gene, c-Mpl.22,23 In each of these syndromes, severe thrombocytopenia will persist beyond the neonatal period, although TAR patients usually show partial normalizations of platelet count over the first few years of life.23 On the other hand, CAMT patients do not exhibit improved platelet counts over time, with many progressing to bone marrow failure.24
Thrombocytopenia 2 (THC2)
Also known as ankyrin repeat domain 26– (ANKRD26)-related thrombocytopenia (ANKRD26-RT), thrombocytopenia 2 is an inherited autosomal-dominant condition mapped to chromosome 10p11.1-p12. THC2 may account for 10% or more of all inherited thrombocytopenias25 and is characterized by moderate thrombocytopenia, dysmegakaryopoiesis, significantly elevated plasma TPO levels, and in a subset of patients, a combination of decreased platelet a-granule content and decreased integrin a2. Twelve different mutations in a very limited region (22 bp) of the 5′-UTR of ANKRD26 have been identified in 21 pedigrees and are considered causative.26 Interestingly, each mutation enhances the transcriptional activity of ANKRD26, leading to the interesting hypothesis that THC2 results from over expression of ANKRD26.
Thrombocytopenia 5 (THC5)
Many families with germline mutations in ETS variant 6 (ETV6) associated with autosomal dominant thrombocytopenia and predisposition to leukemia were recently described.75,76 Bone marrow examination of affected individuals shows small, hypolobulated immature megakaryocytes. Thrombocytopenia is highly penetrant exhibited by almost 100% of affected individuals. Only a fraction of patients with ETV6 germline mutations develop leukemia or other malignancies.
Familial platelet disorder with predisposition to acute myeloid leukemia (FPD/AML)
This is an autosomal dominant disorder characterized by moderate thrombocytopenia, dysmegakaryopoiesis, variable platelet defects, and a propensity to develop myelodysplastic syndromes and/or AML.27,28 Germ-line mutations or deletions in Runt Related Transcription Factor 1 (RUNX1) are thought to be causative, but the precise mechanisms whereby RUNX1 regulates megakaryopoiesis, platelet formation and platelet function are not completely understood.
In FPD/AML, myeloproliferative leukemia protein (MPL) and platelet factor 4 (PF4) expression levels are decreased.29-31 Several heterozygous mutations of RUNX1 have been identified as causative in FPD/AML.28,32 Collectively, these findings point to the importance of RUNX1 in normal megakaryocyte differentiation.
Macrothrombocytopenia (large platelets with thrombocytopenia)
Immune thrombocytopenia (ITP) Immune thrombocytopenia (ITP) is the most common acquired cause of thrombocytopenia with large platelets, particularly in children.33 Patients typically present with a history of significant, acute onset mucocutaneous bleeding. The platelets are large as a result of increased turnover due to rapid consumption and autoantibody mediated destruction in the spleen. Importantly, when children present with “ITP” that is refractory to treatment and/or there is a family history of thrombocytopenia, an alternative diagnosis of inherited, congenital thrombocytopenia should be considered33 (see XLT, MYH9, and inherited thrombocytopenia syndromes).
Platelet-type or type 2B von Willebrand disease (vWD)
Chronic thrombocytopenia with large platelets is also typical of platelet-type or type 2B VWD.34-36 An assessment of von Willebrand factor (VWF) antigen and activity as well as low-dose ristocetin-induced platelet aggregation can establish this diagnosis. Mutation analysis of VWF and GPIb can identify genetic variants responsible for this disorder.37,38
May-Hegglin anomaly, and Fechtner, Sebastian, and Epstein syndromes
Genetic variants of MYH9, the gene for the heavy chain of nonmuscle myosin IIA (NMMHC-IIA) are responsible for a number of macrothombocytopenias, including May-Hegglin anomaly, and Fechtner, Sebastian, and Epstein syndromes.39 Other indicators include the following:
- The presence of Dohle-like bodies in leukocytes on the peripheral blood smear, which can be readily detected by immunofluorescence-based detection of aggregates of NMMHC-IIA
- A family history of associated nephritis, hearing loss, or cataracts (although the manifestation of these defects may be subtle in children, requiring formal audiometry for early detection)40
Gray platelet syndrome (GPS)
Large, pale platelets on the peripheral blood smear is often a characteristic finding in the gray platelet syndrome (GPS).6 Supportive findings would be decreased platelet aggregation with collagen and/or thrombin and the absence of mature alpha granules by electron microscopy. Exome and RNA sequencing of twenty patients by three independent groups led to the discovery that mutation in the neurobeachin-like protein 2 gene (NBEAL2) is the cause of GPS.41-43
Bernard Soulier syndrome (BSS)
Bernard Soulier syndrome is characterized by large, granular platelets on smear, moderate to severe bleeding, and thrombocytopenia.6 The diagnosis can be established by assessment of the surface expression of the GPIb/IX/V complex by flow cytometry.
An autosomal-recessive inheritance is typical, with asymptomatic heterozygous carriers. However, autosomal-dominant mutations resulting in a mild phenotype have also been reported and may be the most common cause of mild macrothrombocytopenia.44-46 The detection of genetic variants of GPIba, GPIbb and GPIX genes is clinically available and can serve to establish the diagnosis and assist in genetic counseling.
Paris Trousseau/Jacobsen syndrome (PT/J)
This condition results from a genetic abnormality in friend leukemia integration 1 transcription factor (Fli1), a transcription factor important in megakaryopoiesis. Neonates with PT/J syndrome exhibit associated cardiac defects, developmental delays, and giant platelets with large alpha granules visible by electron microscopy.47,48
Mediterranean macrothrombocytopenia
Mediterranean macrothrombocytopenia is characterized by mild thrombocytopenia and the absence of clinically significant bleeding and has been reported to result from heterozygosity for genetic variants of the GPIb/IX/V complex.49
DiGeorge or velo-cardio-facial syndromes
The 22q deletion syndrome (22qDS), also known as DiGeorge or velo-cardio-facial syndromes, is relatively common, affecting at least 1 in 4000 live births.50 The syndrome is characterized by decreased platelet count, increased mean platelet volume, and decreased expression of platelet surface GPIb/IX/V, with variable associated cardiac anomalies, hypocalcemia, and immune defects. However, these symptoms may be subtle, and an accurate diagnosis should be made by the finding of genetic variants of the T-box 1 (TBX1) and glycoprotein 1b platelet subunit B (GP1BB) genes in children and parents.51,52
Storage pool disease (SPD)
Storage pool disease (SPD) is a heterogeneous group of inherited platelet function disorders in which selective defects in platelet aggregation result from an inability to store and/or release the content of dense granules and/or a granules.6 In general, the platelets fall within the normal range for size on most cell counters.
In storage pool disease affecting dense granules, the dense granules are storage sites for serotonin, ADP and ATP, key mediators of enhanced platelet activation. SPD affecting dense granules is more common and includes δ-SPD, aδ-SPD, Hermansky-Pudlak syndrome (HPS), Chediak-Higashi syndrome (CHS) and Griscelli syndrome.
Isolated δ-SPD is characterized by easy bruising, mucocutaneous bleeding, and excessive postoperative and postpartum hemorrhage.53 The platelets are morphologically normal on Wright-stained smears, and the diagnosis must be made by the finding of decreased dense granule constituents and/or EM documentation of an absence of dense granule-limiting membranes and contents. The most consistent finding is that adenine nucleotides are reduced with an increased ATP/ADP ratio and normal lysosomal enzyme levels. Evidence for autosomal dominant inheritance has been obtained,54 but the genetic basis has not yet been defined.
In combined aδ-SPD, which is much less common than isolated δ-SPD, δ-granules and/or their contents are uniformly decreased, with a variable deficiency of a-granules and/or their constituents.53 Platelets in these patients form significantly smaller thrombi in flowing blood than those seen with platelets from patients with δ-SPD. The mode of inheritance appears to be autosomal dominant,54 but the genetic basis has not been defined.
Storage pool disease affecting a-granules
SPD affecting solely a-granules (a-SPD) is the rarer subgroup and includes Gray platelet syndrome (see above) and Quebec platelet disorder.
Quebec platelet disorder (QPD) is an extremely rare, autosomal dominant disorder that was originally identified by the finding of low levels of platelet a-granule factor V but normal plasma factor V.53,55 QPD is characterized by protease-related degradation of many platelet a-granule proteins, including P-selectin. One laboratory finding is defective procoagulant activity resulting from defective formation of the prothrombinase complex. Patients with QPD express a tandem duplication of a 78-kb genomic segment including the urokinase plasminogen activator gene (PLAU) and its regulatory elements, implicating this gene in the cause of the disorder.56
Hermansky-Pudlak syndrome (HPS) and Chediak-Higashi syndrome (CHS) include abnormalities of other lysosome-related organelles and thus present with melanosomal defects, which cause a lack of pigmentation of the skin and hair.53,54 Several genes have been associated with HPS, being HPS1 the most commonly affected and other less common such as AP3B1; HPS3; HPS4; HPS5; HPS6; DTNBP, BLOC1S3; AP3D1, causing genetic defects that disrupt organelle biosynthesis and protein trafficking. In CHS, characteristic symptoms are bleeding, giant inclusion bodies in cells, severe immunologic defects, progressive neurological dysfunction, and a lymphoproliferative syndrome. Mutations of the lysosomal trafficking regulator (LYST) gene result in a truncated CHS protein (LYST).
The Griscelli syndrome is inherited in an autosomal recessive pattern and is characterized by unusually light (hypopigmented) skin and light silvery-gray hair starting in infancy. The three types of the syndrome are caused by mutations in three different genes. Type 1 is caused by mutations in the myosin VA (MYO5A) gene and is associated with severely impaired brain function. Type 2, which appears to be the most common form, is caused by mutations in RAB27A, a gene for a small GTPase involved in vesicular transport and organelle dynamics.57,58 Affected individuals have immune system abnormalities as well as hypopigmentation. Additionally, in these patients, lymph nodes and other organs (including the brain) become infiltrated by activated T-cells and macrophages, which phagocytize blood cells (known as hemophagocytosis). These individuals can exhibit neurologic symptoms due to brain infiltration by the activated hematopoietic cells (hemophagocytic syndrome). Type 3 results from mutations in the melanophilin (MLPH) gene and is associated with only hypopigmentation; affected individuals do not exhibit impairment of brain function or immune system abnormalities (US National Library of Medicine. Genetics Home Reference. Available at: http://ghr.nlm.nih.gov/condition/griscelli-syndrome).
Next

{kind=link}