Congenital Platelet Function Disorders
Disease Overview
Defects in platelet function comprise a heterogeneous group of disorders with bleeding symptoms ranging from mild bruising to severe mucocutaneous hemorrhage. Platelet function disorders can be classified according to their relationship to one or more of the stages of platelet thrombus formation, each of which is mediated by specific platelet membrane receptors. Rare genetic variants of these receptor genes can cause defects in platelet adhesion, activation, granule release, prothrombinase activity, aggregation, or clot retraction.
Many of these disorders share common treatments, but some therapies have greater efficacy for one patient relative to another and should be individualized to provide optimal symptom control. Currently, efforts are concentrated on developing methods to rapidly and accurately diagnose patients with platelet function disorders, taking advantage of accumulated and curated databases of genetic variants in candidate genes, and to improve therapy to prevent life-threatening bleeding.
Normal Platelet Function
Platelets initiate hemostasis and stable vascular repair by binding to specific constituents of the subendothelial matrix, particularly collagen, at sites of vascular damage. This process leads to the recruitment of other cellular and plasma constituents that facilitate the repair process. Defects in the normal schema can result in a bleeding syndrome.
Because platelets play a central role in the maintenance of hemostasis, genotypic variations can result in significant phenotypic alterations, increasing the risk for morbidity or death from excessive bleeding or thrombosis.1,2
This summary describes the participation of platelets in normal hemostasis and the effects of the former rare gene variants that are associated with significant hemostatic perturbations and platelet dysfunction. The process of platelet thrombus formation is shown in Figures 1-3. The most common inherited platelet function disorders are listed in Figure 4.
Initiation: Platelet Adhesion
The participation of platelets begins during the transient, and to a certain extent reversible, initiation phase, wherein the A1 domain of plasma von Willebrand factor (VWF) binds to exposed collagen fibrils within the subendothelial matrix. This initial interaction has been described as a rolling contact between platelets and the exposed VWF-collagen interface. With VWF binding to collagen, a structural change in VWF immediately ensues, rendering it a ligand for the platelet membrane receptor, the glycoprotein (GP) Ib complex (Figure 1). This complex is a heptamer composed of two GPIba subunits, two GPIbb subunits, two GPIX subunits and one GPV subunit, each of which is coded by the distinct genes, GP1BA, GP1BB, GP9 and GP5, respectively.
Rare variants of the GP1BA or GP1BB genes have been shown to cause the Bernard-Soulier syndrome (BSS), characterized by macrothrombocytopenia and defective platelet adhesion to collagen.
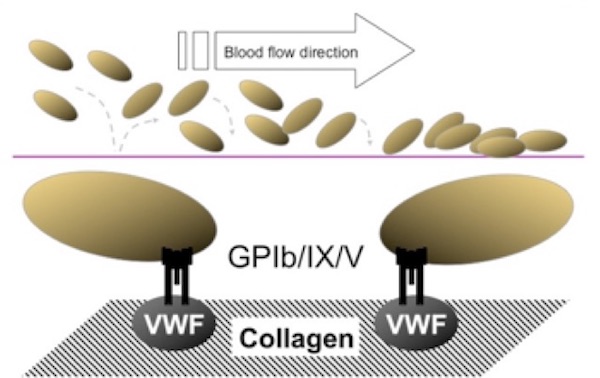
Figure 1. Initiation (adhesion). Initiation of platelet adhesion to collagen-bound von Willebrand Factor (VWF) is mediated by the GPIb/IX/V membrane receptor complex.
(Figure adapted from Simon D, Kunicki T, Nugent D. Platelet function defects. Haemophilia. 2008;14:1240–1249).
Extension: Platelet Activation/Secretion
Extension is the next phase of platelet participation in hemostasis and is represented by additional, stable adhesion and platelet activation, which occur very rapidly and are considered to be essentially concurrent activities (Figure 2). The adherent platelet becomes activated leading to an increased affinity of additional collagen receptors, namely GPVI and the integrin a2b1. These receptors mediate a firmer and persistent attachment of the platelets, forming a more consolidated monolayer on the VWF-collagen interface and markedly enhancing the activation of other adherent platelets. Activated platelets secrete important agonists from both a-granules and dense granules, such as adenosine diphosphate (ADP) and p-selectin, which further propagate platelet activation and recruit additional circulating platelets to become activated and to release granule contents.
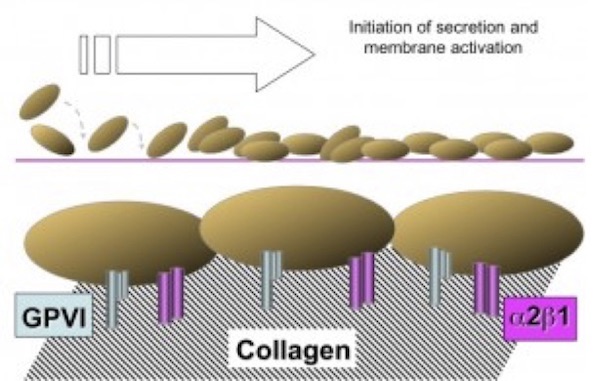
Figure 2. Extension (activation/secretion). Additional adhesion of platelets to collagen mediated by the GPVI and integrin α2β1 receptors (extension) leads to platelet activation, secretion of additional platelet agonists from platelet granule stores, and initiation of prothrombinase activity at the platelet surface.
(Figure adapted from Simon D, Kunicki T, Nugent D. Platelet function defects. Haemophilia. 2008;14:1240–1249).
During the course of this amplification process, the activated platelet produces and/or releases additional agonists, including thromboxane A2 (TXA2), and an internal enzyme scramblase that mediates the flip of phosphatidyl serine to the platelet membrane surface, which serves as a potent catalytic surface for prothrombin conversion to thrombin.
Numerous platelet disorders that involve the extension phase have been described, including defects in GPVI or integrin a2β1, and a diverse group of disorders known as the storage pool defects, which appear to be the most common of these platelet function disorders.
Another defect related to the extension phase is the rare autosomal recessive Scott syndrome.3 Upon activation, Scott syndrome platelets are unable to transport phosphatidyl serine from the inner to the outer phospholipid membrane. Consequently, factor Va–Xa and VIIIa–IXa complexes are unable to bind to the membrane, resulting in decreased thrombin generation and fibrin formation. Mutations in the anoctamin 6 (ANO6) gene that encodes for TMEM16F (transmembrane protein 16F), which is localized in the plasma membrane and confers calcium-dependent scrambling of membrane phospholipids have been described in these patients.3,4
Consolidation: Platelet Cohesion/Aggregation
The next phase of platelet participation is consolidation (Figure 3), wherein the platelets undergo cohesion (or aggregation), mediated by the multivalent binding of plasma fibrinogen (or platelet fibrinogen released from a-granules) or to a lesser extent VWF, to activated integrin aIIbb3 on adjacent platelets. The integrin subunits αIIb and β3 are encoded by the genes ITGA2B and ITGB3, respectively. Rare variants in either gene have been found to cause the classic disorder resulting from defective fibrinogen binding known as Glanzmann thrombasthenia (GT).5
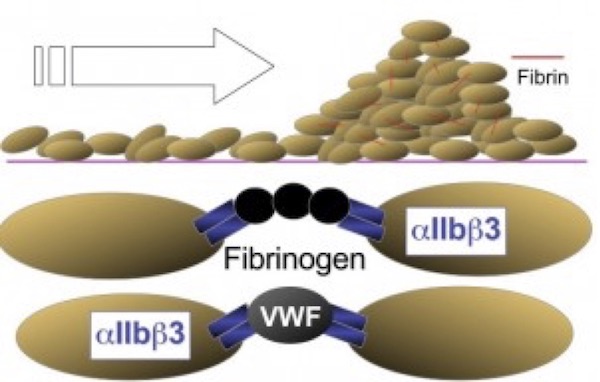
Figure 3. Consolidation (cohesion/aggregation). Consolidation of the nascent thrombus is accomplished by platelet-platelet cohesion (aggregation), mediated by the binding of fibrinogen and/or VWF to the integrin αIIbβ3, and the concurrent development of an intermeshed fibrin network.
(Figure adapted from Simon D, Kunicki T, Nugent D. Platelet function defects. Haemophilia. 2008;14:1240–1249).
The most common platelet function disorders (Figure 4) involve defects in one or more of these important components of the platelet activation process.
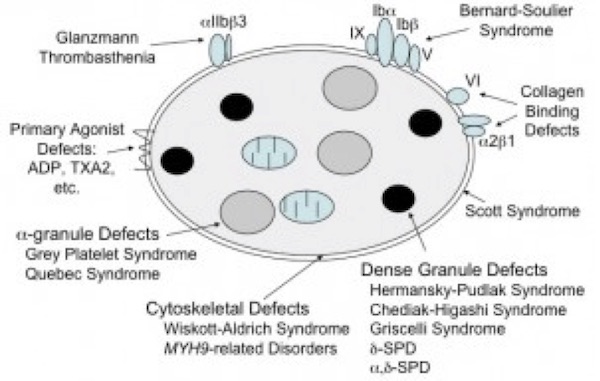
Figure 4. Most common inherited platelet defects.
ADP, adenosine diphosphate; αδ-SPD, combined α-granule and δ-granule storage pool disease; δ-SPD, isolated δ-granule storage pool disease; MYH9, myosin heavy chain 9 gene; TXA2, thromboxane A2.(Figure adapted from Simon D, Kunicki T, Nugent D. Platelet function defects. Haemophilia. 2008;14:1240–1249).
Additional Factors that Influence Hemostasis
The Impact of ThrombocytopeniaMegakaryocytes produce ~100 billion platelets daily to maintain a normal range of circulating platelets of 150-400,000 platelets per microliter (mL) of blood. Thrombocytopenia, whether inherited or acquired, can have an influence on all phases of platelet thrombus formation. In a healthy individual, significant mucocutaneous bleeding normally will likely not be observed until the platelet count falls below 20,000–30,000 /mL. In states of high platelet turnover (compensated thrombopoiesis), such as immune-mediated thrombocytopenia, the platelets are generally younger, slightly larger and more hemostatically active, so that serious bleeding may not be observed until the platelet count falls to <15,000 /mL.
Other defects that warrant special consideration, such as the MYH9-related giant platelet disorders and Wiskott-Aldrich Syndrome (WAS), are discussed in greater detail in the Platelet Defects section.
Table 1 summarizes most known congenital platelet disorders, mode of inheritance, some key diagnostic laboratory findings, the involved protein/receptor defects, and the known causative gene variants that causes them. For the definition of the disorder OMIM (Online Mendelian Inheritance in Man) definitions were used.
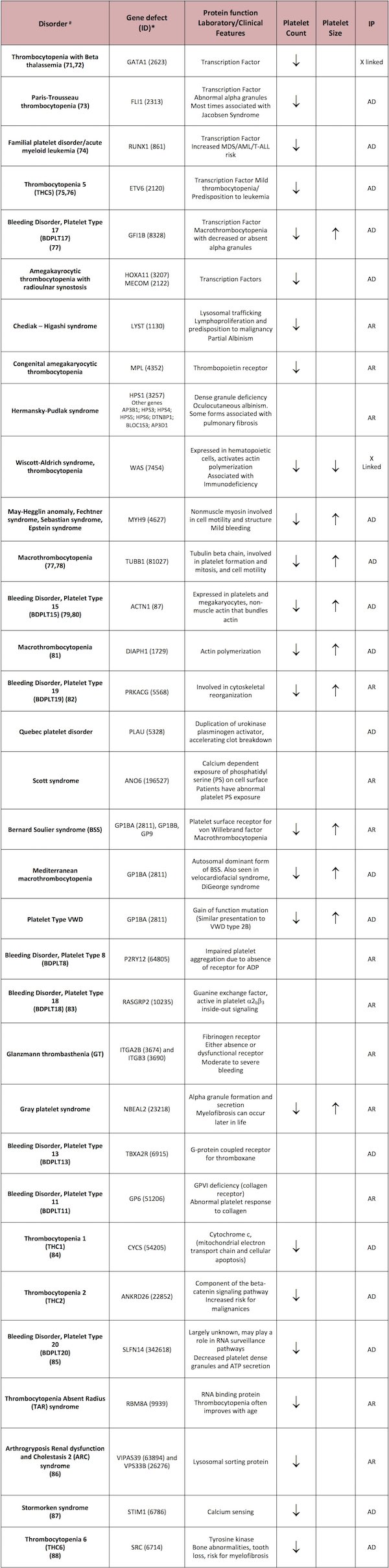
IP Inheritance Pattern
# Many disorders (and their reference) have the official OMIM site name https://www.omim.org
* https://www.ncbi.nlm.nih.gov/gene
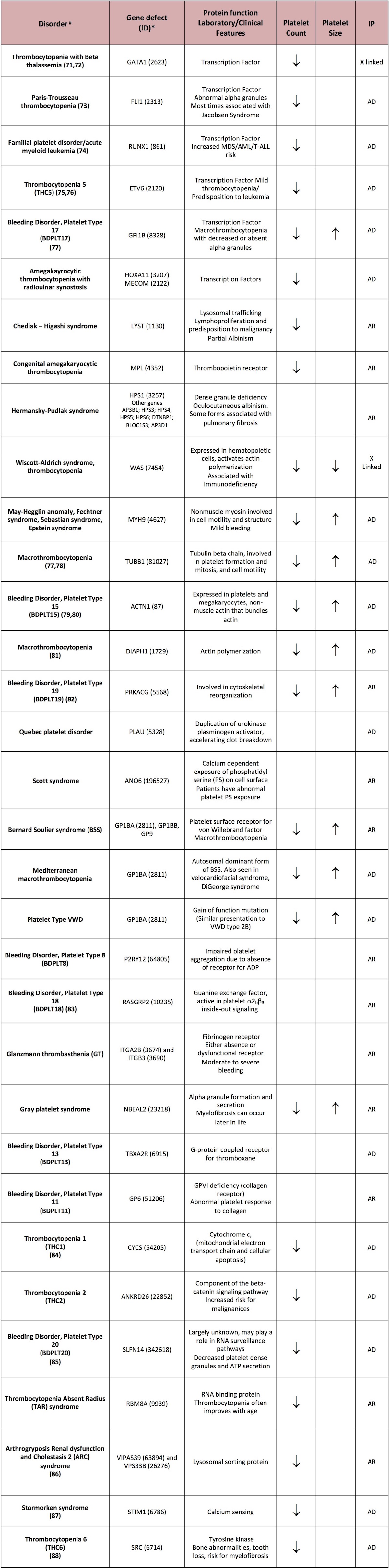
Platelet function disorders are not commonly severe; some argue that these may be underdiagnosed, but some of the conditions occur in a limited number of kindreds worldwide, being more frequent where consanguinity is prevalent or in geographically or ethnically isolated communities. Among the more common secretory defects, compound heterozygous mutations predominate, with autosomal dominant inheritance being rare. Age of onset and family history are crucial factors that can aid in the diagnostic evaluation and choice of therapy.
