Congenital Deficiency of Vitamin K-Dependent Clotting Factors
Medications/Treatment
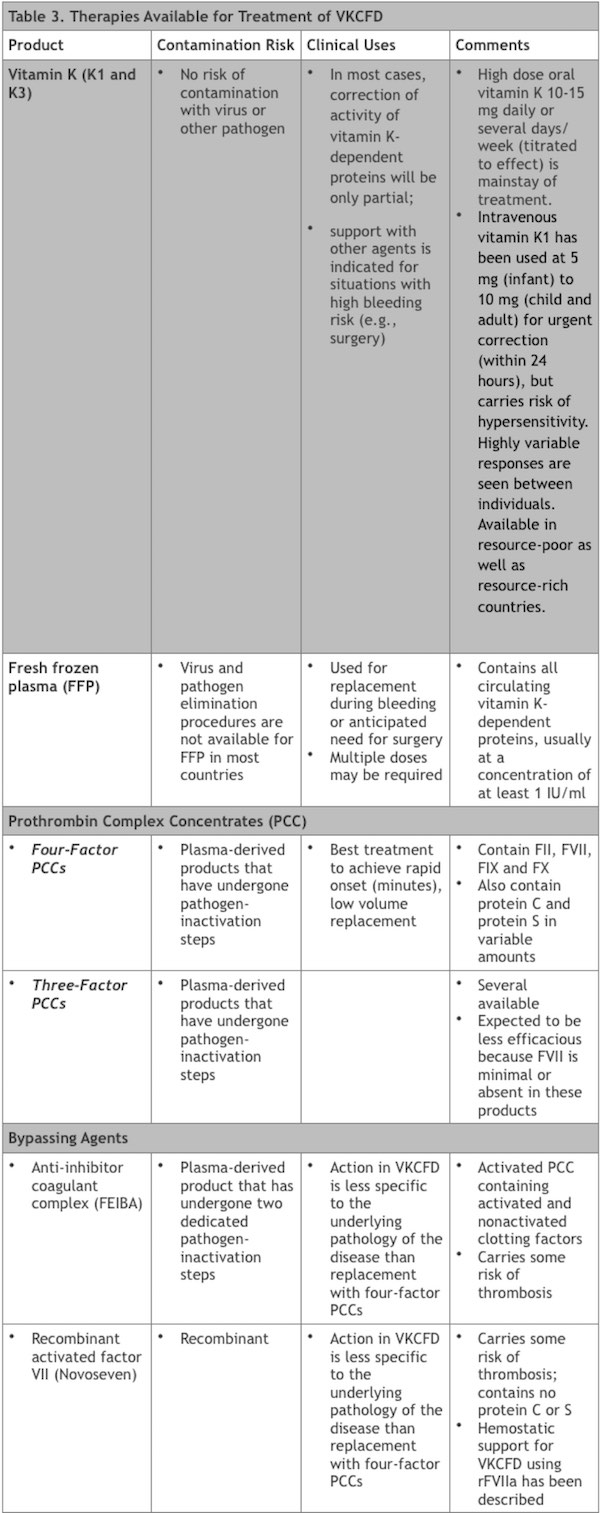
The most important maintenance medication for the treatment of VKCFD1 and VKCFD2 is vitamin K, while several additional therapies are available for supportive care and prophylaxis for surgery or anticipated bleeding risk.
Vitamin K. (Vitamin K1; vitamin K3 may also be efficacious) Daily high dose oral vitamin K is the mainstay of treatment. Parenteral (intravenous or subcutaneous) vitamin K is also effective for supportive care. This therapy is available in resource-poor as well as resource-rich countries and carries no risk of contamination with virus or other pathogens. In most cases, correction of activity of vitamin K-dependent proteins will be partial and support with other agents is indicated for situations with high bleeding risk.
Fresh frozen plasma (FFP). FFP contains all of the circulating vitamin K-dependent proteins, usually at a concentration of at least 1 IU/ml. Therefore FFP can be used for replacement during bleeding or anticipated need for surgery, although multiple doses may be required. FFP with virus and pathogen elimination procedures is the preferred product but is not uniformly available worldwide.
Prothrombin complex concentrates (PCCs). Availability of PCCs has been variable in the past; however, recognition of their efficacy in the reversal of warfarin anticoagulation in individuals without VKCFD has increased the profile of these drugs where available.
- Four-factor PCCs: contain FII, FVII, FIX and FX and are the best treatment to achieve rapid onset (minutes), low volume replacement using a product that has undergone pathogen-inactivation steps.41 These products also contain protein C and protein S in variable amounts.
- Three-factor PCCs: it is worth noting that several three-factor PCCs have been available and would be expected to be less efficacious because FVII is minimal or absent in these products.
Bypassing agents. These products contain activated clotting factors and are available in many coagulation centers. These include the activated PCC anti-inhibitor coagulant complex (Factor VIII Inhibitor Bypassing Activity [FEIBA], Baxter) and the recombinant activated factor VII (rFVIIa [Novoseven], Novo Nordisk). Each of these agents carries some risk of thrombosis and their action in VKCFD is less specific to the underlying pathology of the disease than replacement with four-factor PCCs; successful hemostatic support in a case of VKCFD using rFVIIa has been described.33
The therapies used for management of individuals with VKCFD are summarized in Table 3.
Individuals with VKCFD should receive vitamin K. Although a single case report has implicated VKCFD as potentially associated with a thrombotic event, the report does not establish causation and VKCFD is a disorder that is complicated by bleeding and not by thrombosis. Vitamin K supplementation should not be withheld from any patient. Moreover, oral and subcutaneous administration of vitamin K1 is safe and well tolerated with no risk of harm to the patient from excess doses. Intravenous and intramuscular vitamin K1 is reserved for when oral or subcutaneous routes are not feasible as both intravenous and intramuscular administration have been associated with rare hypersensitivity/anaphylaxis-like reactions, including fatalities.42,43,44
For the treatment of VKCFD2 typical doses for neonates and children are 1-5 mg/day; 10 mg/day dosing in adults has improved clotting factor into or near the normal range in most reports.8 Moderately rapid (< 24 hours) correction to nearly complete thrombin generating potential has recently been detailed with the use of intravenous dose of 10 mg of vitamin K in VKCFD2 in an illustrative case presentation.39,45 Laboratory monitoring is essential to demonstrate correction of the prothrombin and partial thromboplastin times and factors II, VII, IX and X activity, with titration of dose to effect.
The treatment of VKCFD1 may require higher doses. It is not unusual for doses of 20 mg/day to be required to demonstrate increased activity of factors II, VII, IX and X and some individuals demonstrate additional incremental improved VKDP activity with even higher doses. For individuals with severe VKCFD1, oral therapy generally results in some improvement of bleeding symptoms and partial correction of factor activities. Several GGCX mutations have been expressed in in vitro systems, with the demonstration that the expressed variant GGCX proteins having decreased binding affinity for vitamin K.30 Supplementation of vitamin K in excess may drive more favorable kinetics of the vitamin K binding by carboxylase, thus explaining the therapeutic effect of vitamin K in individuals who have these types of mutations. In this scenario, the excess of circulating undercarboxylated prozymogen proteins is a substrate pool that can be carboxylated rapidly after supplementing vitamin K in excess. Currently, it is not possible in general to predict a good or bad response to vitamin K supplementation based upon the genotypic spectrum.5 As genotype/phenotype correlations for GGCX mutations are better described, specific defects in particular domains of the GGCX protein that correspond to carboxylation of specific proteins, as well as amelioration with vitamin K supplementation, may become apparent, as discussed with illustrative cases in the section “Pattern of Inheritance.”
Fresh frozen plasma and /or four-factor PCCs provide replacement with fully carboxylated coagulation proteins for emergency bleeding or for surgery. Multiple doses of plasma may be required, with the concomitant risk of volume overload.
For additional information on the appropriate use of PCCs and bypassing agents in individuals with VKCFD, see Medications.
Prognosis and Life Course
VKCFD may present with severe neonatal bleeding, and the prognosis in the case of perinatal intracranial hemorrhage may be guarded. If early intracranial hemorrhage is avoided, management of severe VKCFD including (most importantly) regular maintenance supplementation of vitamin K may result in mild to moderate levels of biochemical abnormalities, favorable outcomes and good quality of life. This is the norm for individuals with mild VKCFD managed with regular oral vitamin K replacement.
Special management, including high-risk obstetric care and careful vitamin K replacement are recommended for the pregnant woman with VKCFD. An illustrative lesson of a life course and anticipatory management of VKCFD can be gained through study of the original case report and subsequent published reports of the subject’s childhood and adult obstetrical history. 6,7,45,46
What is less clear at the present time is the best approach to surveillance and management of potential manifestations of VKDP undercarboxylation outside of the coagulation system (i.e. MGP, osteocalcin, Gas6, etc); it appears that symptoms involving dermatologic, opthalmologic, cardiac and skeletal symptoms may be common in individuals manifesting VKCFD1, although not in VKCFD2. Dermatologic involvement is particularly common in individuals with GGCX mutations, although the phenotypic expression of the GGCX mutations in one extended kindred included PXE-like skin and opthalmologic abnormalities without coagulation factor deficiencies.36 This raises the possibility that VKCFD1, PXE-like disorder with VKCFD, and PXE-like disorder without coagulation factor deficiency may represent an overlapping disease spectrum, in which different GGCX domain mutations differentially inactivate specific involved VKDPs. What is clear is that the age of onset of the abnormal skin phenotype is variable and usually years later than the onset of the coagulation deficiency phenotype. Clinical surveillance for the onset of PXE-like skin findings (cutis laxa, increased skin folds, yellow papules) is warranted throughout childhood and into adult, even in the setting of apparent efficacy of vitamin K supplementation in regards to coagulation protein correction. If skin findings do develop, opthalmologic screening with fundoscopic imaging is advisable to identify angioid streaks, peau d’orange, and pigmentary retinopathy; while the opthamologic anomalies that have been observed in VKCFD1 patients with PXE-like skin findings have not been functionally significant in the documented cases, the number of cases is small and follow up short and comprehensive long-term experience is needed. An evidence-based approach to recommendations in regards to screening for cardiac and bone disease (in the absence of dysmorphology on physical examination) is more difficult. It has been suggested that all individuals with cardiac and bone defects have carried a mutation affecting the HTTM-coding region (AA 56-315 encoding the GGCX transmembrane domains 1-4) on at least one allele, and that individuals carrying mutations in this region may benefit from screening with echocardiogram and bone densitometry.5 Consideration of bone densitometry for all VKCFD1 patients may be justified regardless, given the common epidemiologic finding of low bone density and osteoporosis in individuals with hemophilia A and hemophilia B.
